
Todos los seres humanos tienen 22 pares de cromosomas iguales, denominados autosomas, y un par de cromosomas diferentes según el sexo del individuo, los cromosomas sexuales o heterocromosomas.
Los cromosomas de cada especie poseen una serie de características, como la forma, el tamaño, la posición del centrómero y las bandas que presentan al teñirse. Este conjunto de particularidades, que permite identificar los cromosomas de las distintas especies, recibe el nombre de cariotipo, y su representación gráfica, ordenada por parejas de cromosomas homologos se denomina cariograma.

Los únicos tratamientos que han demostrado una influencia significativa en el desarrollo de los niños con síndrome de Down son los programas de Atención Temprana, orientados a la estimulación precoz del sistema nervioso central durante los seis primeros años de vida. Los individuos con grandes dificultades para el aprendizaje a menudo han sido internados en instituciones, pero se ha comprobado que deben vivir en su domicilio, donde desarrollan de forma más completa todo su potencial. Cuando éste es demasiado protector, los chicos y chicas tienden a dejarse llevar, descubriendo escasamente sus potencialidades. Los contextos estimulantes ayudan a que se generen conductas de superación que impulsan el desarrollo de la inteligencia. 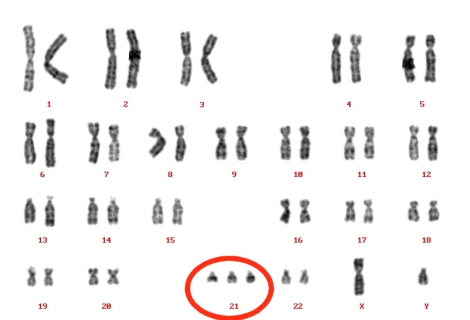

- El ultrasonido puede revelar órganos reproductores femeninos pequeños o subdesarrollados.
- La hormona luteinizante y foliculoestimulante sérica se encuentra elevada.

Sindromes
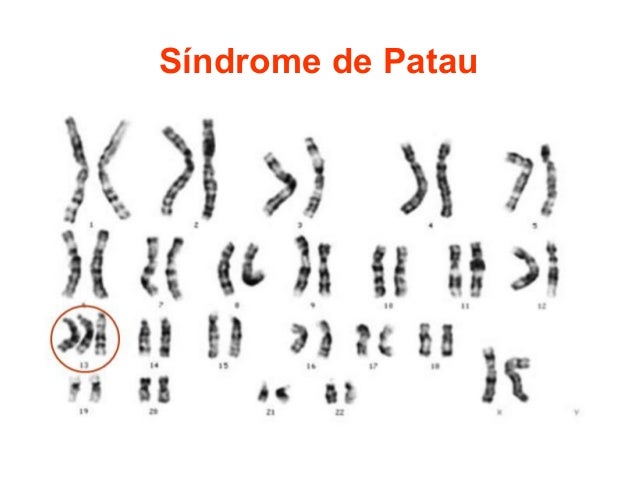
Síndrome de Patau
1. Definición
El síndrome de Patau o trisomía del cromosoma 13 es una
enfermedad cromosómica rara caracterizada por la presencia de un cromosoma 13
adicional. Fue descrita por primera vez por Patau en 1960.
2. Causas
La trisomía 13 puede darse por:
a) Presencia de un cromosoma 13 extra (tercer cromosoma) en todas las células.
b) Mosaicismo, presencia de un cromosoma 13 extra en algunas de las células.
c) Trisomía parcial, presencia de una parte de un cromosoma 13 extra en las células.
La trisomía 13 puede darse por:
a) Presencia de un cromosoma 13 extra (tercer cromosoma) en todas las células.
b) Mosaicismo, presencia de un cromosoma 13 extra en algunas de las células.
c) Trisomía parcial, presencia de una parte de un cromosoma 13 extra en las células.
Es la trisomía más rara con una prevalencia de uno en 12.000
nacidos vivos y predomina ligeramente en el sexo femenino.
Al igual que en otras trisomías de mayor difusión, como en
el síndrome de Down, la edad materna y paterna es un factor de riesgo y en
estos casos suelen ser superiores a los 30 años.
. Diagnóstico y pronóstico
Para diagnosticar este síndrome se puede hacer mediante la ecografía, que consta un retraso en el crecimiento intrauterino y pueden ser detectadas algunas de las anomalías que caracterizan este síndrome, especialmente la holoprosencefalia (ausencia de desarrollo de lo que será el lóbulo frontal del cerebro) y las distintas malformaciones renales, cardiacas y faciales. En los casos en que se sospeche este síndrome será obligada la amniocentesis para dilucidar el cariotipo fetal.
Para diagnosticar este síndrome se puede hacer mediante la ecografía, que consta un retraso en el crecimiento intrauterino y pueden ser detectadas algunas de las anomalías que caracterizan este síndrome, especialmente la holoprosencefalia (ausencia de desarrollo de lo que será el lóbulo frontal del cerebro) y las distintas malformaciones renales, cardiacas y faciales. En los casos en que se sospeche este síndrome será obligada la amniocentesis para dilucidar el cariotipo fetal.
El pronóstico vital es muy grave; la inmensa mayoría de
estos enfermos fallecen de muy pocas semanas de edad, debido a los problemas
cardiorrespiratorios
4. Síntomas
* Retraso del crecimiento
* Labio leporino
* Ojos muy juntos: los ojos realmente pueden fusionarse en
uno
* Polidactilia
* Retardo psicomotor y mental
* Fisura congénita en el iris del ojo (coloboma)
* Cardiopatías congénitas
* Anomalías diafragmáticas, urogenitales y sensoriales
* Disminución del tono muscular
* Microcefalia y micrognacia
* Testículo no descendido (criptorquidia)
* Riñones poliquísticos
5. Tratamiento
5. Tratamiento
Los recien nacidos con trisomía 13 suelen necesitar
asistencia médica desde el mismo momento del nacimiento debido a que 2/3 de los
casos obtienen puntuaciones inferiores a 7 en el test de Apgar al primer
minuto, cifra que desciende a 1/3 a los 5 minutos de vida. Dado que las
anomalías cardiacas representan la causa principal de morbimortalidad, se
plantea el problema ético de si su recuperación quirúrgica está indicado dado
el pésimo pronóstico del cuadro tanto desde el punto de vista físico como
intelectual. Alrededor de 2/3 de los pacienets son dados de alta y precisan de
atención especializada en el domicilio, requiriendo la intervención de un
equipo multidisciplinar. Los padres han de ser previamente entrenados para la
realización de determinadas tareas y maniobras que pueden ser de importancia
vital para la supervivencia del recien nacido.
Síndrome de Down
1. Definición
Es un trastorno genético causado por la presencia de una copia extra del cromosoma 21, en vez de los dos habituales (trisomía del par 21), caracterizado por la presencia de retraso mental y rasgos físicos peculiares. Debe su nombre a John Langdon Haydon Down que fue el primero en describirlo en 1866.
Es un trastorno genético causado por la presencia de una copia extra del cromosoma 21, en vez de los dos habituales (trisomía del par 21), caracterizado por la presencia de retraso mental y rasgos físicos peculiares. Debe su nombre a John Langdon Haydon Down que fue el primero en describirlo en 1866.
Este síndrome es la causa más frecuente de discapacidad
psíquica congénita y afecta aproximadamente uno de cada 800 bebés.
2. Causas
El síndrome de Down es causado por la presencia de material genético extra del cromosoma 21. Este material extra se puede deber por tres razones:
a) Trisomía libre: dada el 95% de los casos, donde ocurre un error durante la primera división meiótica por la disyunción incompleta del material genético de uno de los progenitores.
El síndrome de Down es causado por la presencia de material genético extra del cromosoma 21. Este material extra se puede deber por tres razones:
a) Trisomía libre: dada el 95% de los casos, donde ocurre un error durante la primera división meiótica por la disyunción incompleta del material genético de uno de los progenitores.
b) Translocación: ocurre cuando una parte del
cromosoma 21 se desprende durante la división celular y se adhiere a otro
cromosoma (normalmente a alguno del par 14). Este tipo de accidente en la
división celular es responsable de aproximadamente del 3%-4% de los casos de
síndrome de Down.
c) Mosaicismo: en este caso, el accidente en la división
celular tiene lugar después de la fertilización. Las personas afectadas tienen
algunas células con un cromosoma 21 adicional y otras con la cantidad normal.
Afectan 1%-2% de los casos.
3. Síntomas
La persona afectada puede presentar:
* Retraso mental en diversos grados
* Baja estatura
* Dermatoglifos atípicos
* Diástasis de músulos abdominales
* Disminución del tono muscular
* Braquiocefalia (región occpital plana)
* Paladar ojival
* Manos, orejas y cuello corto
* Puente nasal deprimido
* Cardiopatía congénita
* Mayor riesgo de desarrollar patologías como hipotiroidismo, diabetes, miopía, leucemia, problemas de audición, etc.
4. Epidemiología
* Retraso mental en diversos grados
* Baja estatura
* Dermatoglifos atípicos
* Diástasis de músulos abdominales
* Disminución del tono muscular
* Braquiocefalia (región occpital plana)
* Paladar ojival
* Manos, orejas y cuello corto
* Puente nasal deprimido
* Cardiopatía congénita
* Mayor riesgo de desarrollar patologías como hipotiroidismo, diabetes, miopía, leucemia, problemas de audición, etc.
4. Epidemiología
La incidencia global del síndrome de Down se aproxima a uno
de cada 700 nacimientos, pero el riesgo varía con la edad de la madre. La
incidencia en madres de 25 años es de 1 por cada 2000 nacidos vivos, mientras
que en madres de 35 años es de 1 por cada 200 nacimientos y de 1 por cada 40 en
las mujeres mayores de 40 años. Por este motivo se recomiendan técnicas de
diagnóstico prenatal a todas las mujeres a partir de los 35 años.
La probabilidad de tener un hijo con síndrome de Down es
mayor a la media para aquellos padres que ya han tenido otro previamente.
Típicamente la probabilidad de tener otro hijo con el síndrome en cada embarazo
subsiguiente es de una por cada cien recién nacidos vivos. Los antecedentes
familiares igualmente incrementan ese riesgo.
Los varones con síndrome de Down se consideran estériles, pero las mujeres conservan con frecuencia su capacidad reproductiva. En su caso también se incrementa la probabilidad de engendrar hijos con el síndrome hasta un 50%, aunque pueden tener hijos sin trisomía.
Los varones con síndrome de Down se consideran estériles, pero las mujeres conservan con frecuencia su capacidad reproductiva. En su caso también se incrementa la probabilidad de engendrar hijos con el síndrome hasta un 50%, aunque pueden tener hijos sin trisomía.
4. Diagnóstico
Existen varias formas de determinar el síndrome de Down, entre ellas están la “triple prueba”, la más común y más utilizada, consiste en pruebas de AFP (Alfa-fetoproteína), estriol y hCG (Gonadotropina coriónica humana) para la detección de niveles anormales que indiquen el padecimiento del síndrome.
Otras formas de diagnosticar el síndrome de Down están ecografías (longitud del fémur, grosor del pliegue nucal, etc.), conteo cromosómico a través de alguna célula fetal conseguida con técnicas como amniocentesis y biopsia de vellosidades coriónicas, etc.
Existen varias formas de determinar el síndrome de Down, entre ellas están la “triple prueba”, la más común y más utilizada, consiste en pruebas de AFP (Alfa-fetoproteína), estriol y hCG (Gonadotropina coriónica humana) para la detección de niveles anormales que indiquen el padecimiento del síndrome.
Otras formas de diagnosticar el síndrome de Down están ecografías (longitud del fémur, grosor del pliegue nucal, etc.), conteo cromosómico a través de alguna célula fetal conseguida con técnicas como amniocentesis y biopsia de vellosidades coriónicas, etc.
5. Tratamiento
La mejoría en los tratamientos de las enfermedades asociadas
al síndrome de Down ha aumentado la esperanza de vida de estas personas, entre
los 50 años y los 60 años. A lo largo de los últimos años se han postulado
diferentes tratamientos prácticos (hormona tiroidea, hormona del crecimiento,
complejos vitamínicos y minerales, etc.) sin que ninguno haya demostrado ningún
efecto positivo significativo en el desarrollo motor, social, intelectual de
las personas con el síndrome de Down.
Los únicos tratamientos que han demostrado una influencia significativa en el desarrollo de los niños con síndrome de Down son los programas de Atención Temprana, orientados a la estimulación precoz del sistema nervioso central durante los seis primeros años de vida. Los individuos con grandes dificultades para el aprendizaje a menudo han sido internados en instituciones, pero se ha comprobado que deben vivir en su domicilio, donde desarrollan de forma más completa todo su potencial. Cuando éste es demasiado protector, los chicos y chicas tienden a dejarse llevar, descubriendo escasamente sus potencialidades. Los contextos estimulantes ayudan a que se generen conductas de superación que impulsan el desarrollo de la inteligencia.
Síndrome de Cri-du-chat
1. Definición
El síndrome del maullido de gato (del francés Cri-du-Chat), es una enfermedad congénita infrecuente con alteración cromosómica provocada por un tipo de deleción estructural del brazo corto del cromosoma 5, caracterizada por un llanto que se asemeja al maullido de un gato.
El síndrome del maullido fue descrito inicialmente por Lejeune en 1963. Tiene una prevalencia estimada de aproximadamente de 1 en 20.000 nacimientos y predomina en las niñas
El síndrome del maullido de gato (del francés Cri-du-Chat), es una enfermedad congénita infrecuente con alteración cromosómica provocada por un tipo de deleción estructural del brazo corto del cromosoma 5, caracterizada por un llanto que se asemeja al maullido de un gato.
El síndrome del maullido fue descrito inicialmente por Lejeune en 1963. Tiene una prevalencia estimada de aproximadamente de 1 en 20.000 nacimientos y predomina en las niñas
2. Causas
La causa del síndrome del maullido del gato es la supresión de genes en el cromosoma 5. Uno de los genes suprimidos llamado telomerasa transcriptasa inversa (TERT) y dependiendo del tamaño de la porción perdida la afección será mayor o menor. La causa de esta rara supresión cromosómica se desconoce, pero se cree que entre el 85%-90% de los casos se debe a la pérdida espontánea de una parte del cromosoma 5 durante el desarrollo de un óvulo o de un espermatozoide. En un 10%-15% de estos casos se debe a que uno de los padres es portador de una reorganización del cromosoma 5 denominada translocación.
3. Síntomas
* La cara suele ser redondeada, llena y mofletuda, con el paladar elevado y escarpado.
* Cabeza pequeña (microcefalia).
* Inclinación de los ojos hacia abajo
* Ojos separados (hipertelorismo).
* Orejas de implantación baja (pueden estar malformadas).
* Quijada pequeña (micrognatia).
* Mala oclusión dental.
* Llanto de tono alto similar al de un gato, debido al anormal desarrollo de la glotis y laringe.
* Bajo peso al nacer y crecimiento lento.
* Un solo pliegue en la palma de la mano (pliegue simiesco)
* Desarrollo lento o incompleto de las habilidades motoras.
* Retraso mental.
* Miopía y atrofia óptica
* Pies planos.
* Metacarpianos y metatarsianos pequeños.
* Pliegue del epicanto (pliegue extra de piel sobre el ángulo interior del ojo)
4. Tratamiento
La causa del síndrome del maullido del gato es la supresión de genes en el cromosoma 5. Uno de los genes suprimidos llamado telomerasa transcriptasa inversa (TERT) y dependiendo del tamaño de la porción perdida la afección será mayor o menor. La causa de esta rara supresión cromosómica se desconoce, pero se cree que entre el 85%-90% de los casos se debe a la pérdida espontánea de una parte del cromosoma 5 durante el desarrollo de un óvulo o de un espermatozoide. En un 10%-15% de estos casos se debe a que uno de los padres es portador de una reorganización del cromosoma 5 denominada translocación.
3. Síntomas
* La cara suele ser redondeada, llena y mofletuda, con el paladar elevado y escarpado.
* Cabeza pequeña (microcefalia).
* Inclinación de los ojos hacia abajo
* Ojos separados (hipertelorismo).
* Orejas de implantación baja (pueden estar malformadas).
* Quijada pequeña (micrognatia).
* Mala oclusión dental.
* Llanto de tono alto similar al de un gato, debido al anormal desarrollo de la glotis y laringe.
* Bajo peso al nacer y crecimiento lento.
* Un solo pliegue en la palma de la mano (pliegue simiesco)
* Desarrollo lento o incompleto de las habilidades motoras.
* Retraso mental.
* Miopía y atrofia óptica
* Pies planos.
* Metacarpianos y metatarsianos pequeños.
* Pliegue del epicanto (pliegue extra de piel sobre el ángulo interior del ojo)
4. Tratamiento
No hay un tratamiento específico disponible para este
síndrome. Se debe abordar el retardo mental y se recomienda la asesoría para
los padres.
Los padres de un niño con este síndrome deben buscar asesoría genética y someterse a una prueba de cariotipo con el fin de determinar si uno de ellos tiene una reordenación del cromosoma 5
5. Pronóstico
El pronóstico varía, pero lo usual es el retardo mental. La mitad de los niños afectados aprende habilidades verbales suficientes para comunicarse, el llanto similar a un gato se vuelve menos evidente a medida que pasa el tiempo, los cambios en la pubertad serán típicos, etc.
Pueden presentar complicaciones como incapacidad de valerse por sí solo e incapacidad de desenvolverse socialmente.
Los padres de un niño con este síndrome deben buscar asesoría genética y someterse a una prueba de cariotipo con el fin de determinar si uno de ellos tiene una reordenación del cromosoma 5
5. Pronóstico
El pronóstico varía, pero lo usual es el retardo mental. La mitad de los niños afectados aprende habilidades verbales suficientes para comunicarse, el llanto similar a un gato se vuelve menos evidente a medida que pasa el tiempo, los cambios en la pubertad serán típicos, etc.
Pueden presentar complicaciones como incapacidad de valerse por sí solo e incapacidad de desenvolverse socialmente.
Síndrome de Turner
1. Definición
El síndrome de Turner es una enfermedad genética caracterizada por presencia de un solo cromosoma X. Fenotípicamente son mujeres (por ausencia de cromosoma Y). Este trastorno inhibe el desarrollo sexual y causa infertilidad. Su incidencia es de alrededor de 1 en cada 2.500 niñas.
Usualmente es esporádico, lo que significa que no es heredado de uno de los padres. En pocos casos, uno de los padres lleva cromosomas reorganizados que pueden ocasionar el síndrome de Turner en una hija; esta es la única situación en la que este síndrome es heredado. La condición se diagnóstica ya sea al nacer o en la pubertad cuando existe ausencia o retraso de la menstruación y se presenta un retraso en el desarrollo de las características sexuales secundarias normales.
El nombre "síndrome de Turner" proviene del médico Dr. Henry Turner, quien fue el primero en describir el conjunto de descubrimientos en 1938. No fue sino hasta 1959 que se identificó la causa del síndrome de Turner (la presencia de un sólo cromosoma X).
2. Síntomas
El síndrome de Turner es una enfermedad genética caracterizada por presencia de un solo cromosoma X. Fenotípicamente son mujeres (por ausencia de cromosoma Y). Este trastorno inhibe el desarrollo sexual y causa infertilidad. Su incidencia es de alrededor de 1 en cada 2.500 niñas.
Usualmente es esporádico, lo que significa que no es heredado de uno de los padres. En pocos casos, uno de los padres lleva cromosomas reorganizados que pueden ocasionar el síndrome de Turner en una hija; esta es la única situación en la que este síndrome es heredado. La condición se diagnóstica ya sea al nacer o en la pubertad cuando existe ausencia o retraso de la menstruación y se presenta un retraso en el desarrollo de las características sexuales secundarias normales.
El nombre "síndrome de Turner" proviene del médico Dr. Henry Turner, quien fue el primero en describir el conjunto de descubrimientos en 1938. No fue sino hasta 1959 que se identificó la causa del síndrome de Turner (la presencia de un sólo cromosoma X).
2. Síntomas
* Talla baja
* Falla gonadal (infertilidad)
* Falla gonadal (infertilidad)
* Micrognatia (falta de desarrollo mandibular)
* Implantación baja del pelo
* Paladar ojival
* Cuarto metacarpiano corto
* Escoliosis
* Rasgos oculares anormales (caída de los párpados)
* Desarrollo óseo anormal
* Implantación baja del pelo
* Paladar ojival
* Cuarto metacarpiano corto
* Escoliosis
* Rasgos oculares anormales (caída de los párpados)
* Desarrollo óseo anormal
* Mamas pequeñas y vello púbico disperso
* Lagrimeo disminuido
* Menstruación ausente
* Pliegue simiesco (un sólo pliegue en la palma)
* Carencia de la humedad normal en la vagina, relaciones sexuales dolorosas
3. Signos y exámenes
* Lagrimeo disminuido
* Menstruación ausente
* Pliegue simiesco (un sólo pliegue en la palma)
* Carencia de la humedad normal en la vagina, relaciones sexuales dolorosas
3. Signos y exámenes
- Cariotipo X0, presente en el síndrome de Turner. El examen
físico revela genitales y mamas subdesarrollados, cuello corto, baja estatura y
desarrollo anormal del tórax.
- El cariotipo muestra 45 cromosomas con un modelo de 44 X,
o es decir, un cromosoma sexual ausente.
- El ultrasonido puede revelar órganos reproductores femeninos pequeños o subdesarrollados.
- El examen ginecológico puede revelar sequedad del
recubrimiento de la vagina.
- La hormona luteinizante y foliculoestimulante sérica se encuentra elevada.
4. Complicaciones
* Anomalías renales
* Presión sanguínea alta (hipertensión)
* Obesidad
* Diabetes mellitus
* Tiroiditis de Hashimoto
* Cataratas
* Artritis
5. Tratamiento
La hormona del crecimiento puede ayudar a una niña con síndrome de Turner a incrementar su estatura. La terapia con reemplazo de estrógenos con frecuencia se inicia cuando la niña tiene 12 ó 13 años de edad y ayuda a estimular el crecimiento de las mamas, del vello púbico y otras características sexuales.
Las mujeres con este síndrome que deseen quedar en embarazo pueden pensar en la utilización de un óvulo de donante.

* Presión sanguínea alta (hipertensión)
* Obesidad
* Diabetes mellitus
* Tiroiditis de Hashimoto
* Cataratas
* Artritis
5. Tratamiento
La hormona del crecimiento puede ayudar a una niña con síndrome de Turner a incrementar su estatura. La terapia con reemplazo de estrógenos con frecuencia se inicia cuando la niña tiene 12 ó 13 años de edad y ayuda a estimular el crecimiento de las mamas, del vello púbico y otras características sexuales.
Las mujeres con este síndrome que deseen quedar en embarazo pueden pensar en la utilización de un óvulo de donante.
No hay comentarios.:
Publicar un comentario